REVIEW Principles of adoptive T cell therapy in cancer Özcan Met1,2,3 & Kasper Mølgaard Jensen1 & Christopher Aled Chamberlain1 & Marco Donia1,2 & Inge Marie Svane1,2 Received: 24 June 2018 /Accepted: 13 August 2018 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 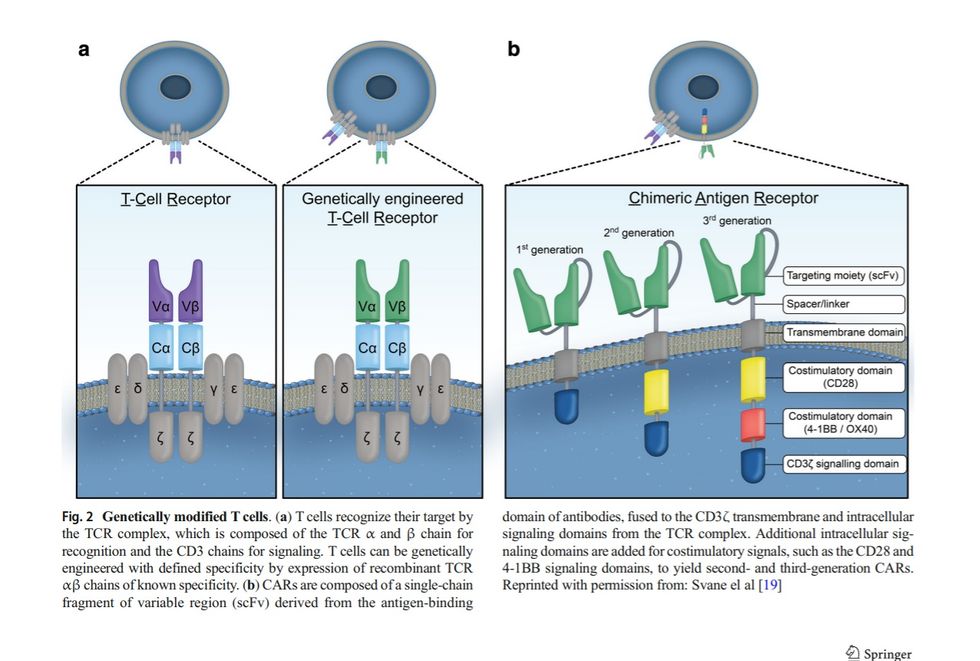
Abstract Adoptive cell therapy (ACT) utilizing either tumor-infiltrating lymphocyte (TIL)-derived T cells or T cells genetically engineered to express tumor recognizing receptors has emerged as a powerful and potentially curative therapy for several cancers. Many ACT-based therapies have recently entered late-phase clinical testing, with several T cell therapies already achieving regulatory approval for the treatment of patients with B cell malignancies. In this review, we briefly outline the principles of adoptively transferred T cells for the treatment of cancer. Keywords Cancer immunotherapy .
Adoptive cell therapy . Tumor-infiltrating lymphocytes . Chimeric antigen receptor . T cells Introduction Cancer immunotherapy is defined as the approach to combatting cancer by generating or augmenting an immune response against cancer cells. Over the past decade, two types of immunotherapy have emerged as particularly effective in cancer treatment: the use of immune checkpoint inhibitors to enhance natural antitumor activity and the administration of specific antitumor immune cells via adoptive cell therapy (ACT). At present, the most widespread type of immunotherapy is the administration of monoclonal antibodies directed against regulatory immune checkpoint molecules that inhibit T cell activation, in particular, cytotoxic T lymphocyte-associated protein-4 (CTLA-4) [1], programmed cell death-1 (PD-1) [2], and programmed death-ligand 1 (PD-L1) [3].
As both a single-agent and in combination, these immune checkpoint inhibitors have demonstrated marked overall and diseasefree survival benefits in multiple clinical trials, paving the way for regulatory approval of these drugs in a variety of solid tumors and hematological malignancies [4–10]. While this treatment modality has been successfully applied in many solid tumors, the main mechanism relies on boosting a pre-existing population of potentially tumorreactive T cells in the patient. Thus, in poorly immunogenic cancer types, immune checkpoint therapy alone is likely to fail [11].
In this regard, the administration of tumor-recognizing T cells via ACT would enable immune-based therapies for these poorly immunogenic cancer types and potentially augment responses in tumors that are already responsive to immune checkpoint therapy. In this review, we present a brief outline of the basic principles of ACT utilizing tumor-infiltrating lymphocytes (TILs) and genetically engineered T cells. ACT modalities The ultimate goal of ACT is to generate a robust immunemediated antitumor response via the infusion of ex vivo manipulated T cells. 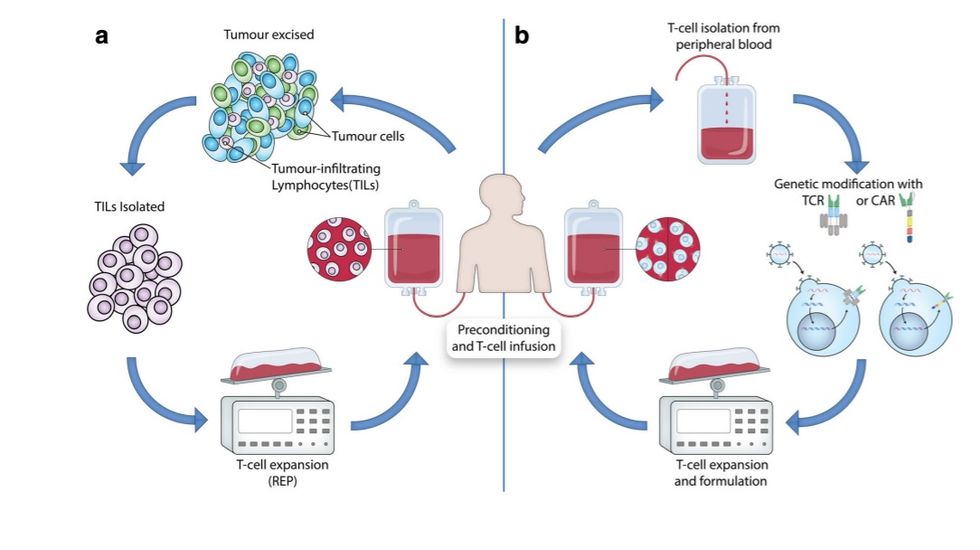
ACT-based strategies utilizing T cells to destroy tumors can be divided into (i) the isolation of naturally occurring tumor-specific T cells from existing tumor masses (TILs), and (ii) the genetic modification of blood-derived T cells to allow for specific recognition of tumor cells. In both settings, T cells are manipulated ex This article is a contribution to the special issue on Anti-cancer Immunotherapy: Breakthroughs and Future Strategies - Guest Editor: Mads Hald Andersen * Özcan Met ozcan.met@regionh.dk 1 Center for Cancer Immune Therapy, Department of Hematology, Copenhagen University Hospital, Entrance 81, Floor 05, 2730 Herlev, Denmark 2 Department of Oncology, Copenhagen University Hospital, Herlev, Denmark 3 Department of Immunology and Microbiology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark Seminars in Immunopathology https://doi.org/10.1007/s00281-018-0703-z
https://doi.org/10.1007/s00281-018-0703-z vivo followed by an expansion and eventual reinfusion back into the lymphodepleted patient (Fig. 1). Naturally occurring tumor-specific T cells TILs are a heterogeneous population of lymphocytes, consisting primarily of T cells and natural killer (NK) cells, that naturally migrate into the tumor and are potentially present in any solid tumor. One of the earliest reports detailing the clinical benefit of lymphocyte infiltration was a case report from 1972 where it was reported that a gastric cancer patient demonstrated total regression of liver metastasis in the absence of prior therapy [12]. The dense infiltration of lymphocytes observed in the resected gastric biopsy suggested the importance of these TILs in curtailing cancer growth. Subsequently, the presence of TILs in tumors has been associated with a favorable prognosis in various cancer types [13–16].
TILs capable of recognizing tumor associated antigens (TAAs) through their endogenous T cell receptors (TCRs) can be isolated from resected tumors; however, the relatively small number of TILs recovered would be inadequate for ACT. The discovery of the T cell growth factor interleukin-2 (IL-2) [17] has allowed the development of a standard method for large-scale in vitro expansion of TILs isolated from patient tumors [18]. This method, involving the exposure of extracted TILs to high dose IL-2 followed by a rapid expansion process utilizing a mixed feeder cell population, was pioneered by Steven Rosenberg and his colleagues at the Surgery Branch of the National Cancer Institute (NCI) and resulted in the production of enough cells for ACT [20]. Initially tested in refractory metastatic melanoma patients, ACT of these cells was found to be an effective treatment option, particularly when preceded by nonmyeloablative lymphodepletion and followed by subsequent high-dose IL-2 treatment [21]. TIL-based ACT relies on (i) nonmyeloablative lymphodepletion, (ii) infusion of large numbers of expanded TILs isolated from a resected tumor, and (iii) IL-2 administration following TIL infusion. The overall approach for growing and administrating TILs is depicted in Fig. 1a. The resected tumor specimen is divided into multiple fragments that are individually grown in IL-2 or enzymatically dispersed into a single-cell suspension. Lymphocytes will then overgrow and typically eradicate tumor cells within 2–3 weeks, resulting in pure TIL cultures. If autologous tumor cells are available, individual TIL cultures can be selected based on attributes such as tumor-reactive interferon-γ (IFN-γ) secretion and cytotoxicity [20]. Selected TIL cultures are then subjected to a rapid expansion protocol (REP) in the presence of excess irradiated feeder cells, an antibody targeting the CD3 complex of the TCR, and high dose IL-2. With this approach, up to 2 × 10^11 lymphocytes can be obtained for infusion into patients [22].
However, difficulties in generating autologous tumor cultures and variations in target tumor quality have prompted many institutions to utilize minimally cultured TILs, where typically all isolated TILs are utilized for further massive expansion and Fig. 1 Different adoptive Tcell transfer (ACT) approaches to harness the immune system to treat cancer (a) Adoptive transfer of anti-tumor T cells isolated from within a patient’s tumor. Tumor-infiltrating T cells (TILs) are extracted from surgically resected tumor samples, then expanded in vitro, followed by re-infusion into the lymphodepleted patient. (b) T cells from patient peripheral blood are isolated and expanded in culture and genetically modified to express either a T cell receptor (TCR) or a chimeric antigen receptor (CAR) that confers the ability to specifically recognize and destroy tumor cells when re-infused into the lymphodepleted patient. Reprinted with permission from: Svane el al [19] Semin Immunopathol infusion [23–25]. The main benefit of this approach is the considerably reduced culture period, which simplifies a significant portion of this complex expansion platform and is less labor-intensive and more cost-effective. Prior to cell infusion, patients are subjected to a preconditioning regimen, commonly including the administration of cyclophosphamide and fludarabine, causing transient host lymphodepletion [26]. This has been shown to increase the persistence of infused TILs, as well as the incidence and duration of clinical responses after TIL therapy [27].
First observed at the NCI, the efficacy of this personalized immunotherapy has been confirmed by multiple independent studies reporting objective response rates of 40 to 50% in patients with metastatic melanoma, including complete tumor regression in 10 to 25% of treated patients (Table 1). The efficacy of adoptive TIL therapy can be put into perspective when considering that metastatic melanoma was a highly lethal neoplasm with only 10% 5-year survival prior to the initiation of TIL therapy [43]. In addition, TIL therapy has mainly been used in late-stage metastatic melanoma cases as a salvage treatment after the failure of standard therapies in patients with multiple metastatic sites. More importantly, the collective experience of various independent studies is that a substantial part of the observed responses are durable, especially in patients achieving complete tumor regression, and that the vast majority of these patients are disease-free many years after treatment [28–30, 44–46]. These findings clearly demonstrate the clinical efficacy of TIL-based ACT and highlight the curative potential of this treatment. For comparison, although objective response rates of around 57% were obtained with combined immune checkpoint blockade in patients with treatment-naive melanoma, the number of patients acquiring complete tumor regression was reported as 2.2%, 8.9%, and 11.5% when treated with antibodies targeting CTLA-4 or PD-1 as a monotherapy, or the combination of both, respectively [6]. Previous reports have shown that prior treatment with IL-2-based immunotherapy and/or anti-CTLA-4 antibodies does not appear to impact the response to adoptive TIL therapy [44, 47]. As PD-1 targeting immune checkpoint therapy has become the standard of care in recent years, we recently investigated whether patients progressing after anti-PD-1 immunotherapy could still respond to an infusion of TILs [22].
We demonstrated that these patients could indeed respond to TIL infusion, and in addition, we found that tumor-reactive T cells heavily infiltrated the tumor microenvironment of patients who had previously failed immune checkpoint treatment. These findings suggest that the mechanisms leading to resistance to current immune checkpoint therapy do not overlap with resistance to TIL-based ACT, and that despite the increasing number of treatment options in metastatic melanoma relegating TIL-based ACT to a third- or fourth-line therapy, the utilization of biomarker-driven strategies to study the tumor of individual melanoma patients failing immune checkpoint therapy can guide future treatment strategies [22]. Whereas treatment-associated mortality is considerably less than that seen with conventional treatments for relapsed or refractory cancers, significant toxicities have been observed in TIL-based ACT. In general, these have been categorized as Common Terminology Criteria for Adverse Events (CTCAE) grade 3 and 4 toxicities and are primarily related to the preconditioning regimen, particularly the administration of highdose IL-2 after cell transfer [44–46]. In this regard, we have previously reported the use of an attenuated IL-2 decrescendo regimen and showed an objective response rate in 10 of 24 (42%) evaluable patients, including three durable complete responders (12%), which is comparable to what has previously been published with ACT plus high-dose bolus IL-2 [30]. Although IL-2-related toxicities were observed, this was generally manageable without requiring intensive care support. The use of attenuated doses of IL-2 may increase the applicability of TIL-based ACT to centers without readily available intensive care units. In addition, due to its high costs in terms of toxicity, it is important to discover predictive criteria for response in order to only expose those patients with a reasonable chance of obtaining a clinical benefit to TIL-based ACT. So far, contrasting results have been reported on the use of tumor mutational burden or tumor neoepitope burden as predictive criteria of response to TIL-based ACT [48]. The observation that melanoma TILs can mediate durable and complete cancer regression in patients with metastatic melanoma has raised considerable interest regarding the possible use of TILs for the treatment of other cancer types. Large-scale TIL growth has been described for a number of solid cancers other than melanoma, including ovarian, breast, colon, cervical, sarcoma, and renal [49–53]; however, only moderate clinical responses have been observed with TILbased ACT. Ongoing research is exploring how to improve the efficacy of TIL-based ACT in melanoma and to extend its efficacy to other common cancers using novel approaches to identify cancer mutations [54, 55], as well as to increase its availability to reference cancer centers. Genetically modified T cells In contrast to TIL-based ACT, the second approach for generating tumor-specific T cell therapies relies on the genetic modification of T cells to enhance antitumor immune function where natural tumor-specific immune responses have failed by manipulating antigen specificity. This is achieved via the transfer of genetic material encoding either a cloned TCR or a Semin Immunopathol synthetic chimeric antigen receptor (CAR) targeting tumor specific antigens. Formed by combining the antigen-binding portions of an antibody molecule with the signaling components of various immunoreceptors and costimulatory molecules, CARs are designed to be highly specific and highly reactive. While many different approaches are utilized to generate genetically modified T cells, the general outline of this approach is depicted in Fig. 1b. Simply, T cells are obtained from peripheral blood, usually after leukapheresis, and activated before being genetically altered and expanded prior to their reinfusion back into the patient.
The patient is often subjected to a preconditioning regimen similar to that of TILbased ACT beforehand. Gene transfer methods commonly used to genetically engineer T cells include the use of transient mRNA transfection [56], retroviral vectors [57], lentiviral vectors [58], transposons [59], or, most recently, homologous recombination after gene editing [60]. Table 1 Clinical responses to different ACT modalities—Tumor-infiltrating lymphocytes (TIL) or genetically-modified T cells (TCR or CAR) Study Disease Type of ACT Antigen target Conditioning
Number of patients Clinical response Rosenberg, 1988 Melanoma TIL Various Cy 20 ORR 11 (55%) (ref. [18]) CR 1 (5%) Dudley, 2005 Melanoma TIL Various Cy + Flu 43 ORR 21 (49%) (ref. [21]) CR 5 (12%) Itzhaki, 2011 Melanoma TIL Various Cy + Flu 31 ORR 15 (48%) (ref. [28]) CR 4 (13%) Radvanyi, 2012 Melanoma TIL Various Cy + Flu 31 ORR 13 (42%) (ref. [29]) CR 2 (6%) Andersen, 2016 Melanoma TIL Various Cy + Flu 25 ORR 10 (42%) (ref. [30]) CR 3 (13%) Morgan, 2006 Melanoma TCR MART-1 Cy + Flu 15 ORR 2 (13%) (ref. [31]) (aa27-35, HLA-A2) Johnson, 2009 Melanoma TCR gp100 Cy + Flu 16 ORR 3 (19%) (ref. [32]) (aa154-162, HLA-A2) CR 1 (6%) Robbins, 2011 Synovial sarcoma TCR NY-ESO-1 Cy + Flu 17 ORR 9 (53%) (ref. [33]) Melanoma (aa157165, HLA-A2) CR 2 (12%) Rapaport, 2015 Multiple Myeloma TCR NY-ESO-1 Other 20 ORR 18 (90%) (ref. [34]) (aa157-165, HLA-A2) CR 16 (80%) Davila, 2014 ALL 2nd CAR CD19 Cy 16 ORR 14 (88%) (ref. [35]) (adult) (CD28) CR 14 (88%) Maude, 2014 ALL 2nd CAR CD19 Cy + Flu 30 ORR 27 (90%) (ref. [36]) (child/young adult) (4-1BB) Other CR 27 (90%) Park, 2018 ALL 2nd CAR CD19 Cy 53 CR 44 (83%) (ref. [37]) (adult) (CD28) Cy + Flu ORR 44 (83%) Maude, 2018 ALL 2nd CAR CD19 Cy + Flu 75 ORR 61 (81%) (ref. [36]) (child/young adult) (4-1BB) CR 61 (81%) Kochenderfer, 2015 NHL/CLL 2nd CAR CD19 Cy + Flu 15 ORR 12 (80%) (ref. [38]) (CD28) CR 8 (53%) Neelapu, 2017 NHL 2nd CAR CD19 Cy + Flu 101 ORR (54%) (ref. [39]) (CD28) CR (54%) Porter, 2015 CLL 2nd CAR CD19 Cy + Flu 14 ORR 8 (58%) (ref. [40]) (4-1BB) Other CR 4 (29%) Turtle, 2017 CLL 3rd CAR CD19 Cy + Flu 24 ORR 16 (67%) (ref. [41]) (CD28/4-1BB) CR 4 (17%) Brudno, 2018 MM 2nd CAR BCMA Cy + Flu 16 ORR 13 (81%) (ref. [42]) (CD28) CR 10 (63%)
These trials make use of different preconditioning regimens (Cy, cyclophosphamide; Flu, fludarabine), and for CAR therapy trials, different signaling elements (CD28 or 4-1BB) are used. For TCR therapy trials target antigen, epitope and HLA-restriction is indicated Semin Immunopathol TCR-modified T cells TCRs are naturally occurring surface receptors on T cells that can recognize peptide antigens presented on the surface of host cells via the major histocompatibility complex (MHC)/ human leukocyte antigen (HLA) system. Genetically modified TCR therapy alters T cell specificity through the expression of a new TCR alpha and beta chain pair that is tumor antigen-specific (Fig. 2a). For this purpose, the TCRs of T cells that can recognize naturally processed and expressed tumor antigens, and therefore specifically attack malignant tissue, have been identified. However, as TCRs bind to peptide/MHC complexes at the cell surface of tumor cells, the tumor-specific TCRs can only be used in a patient population that has this specific MHC or HLA allele. After the isolation and sequencing of a tumor-specific TCR, it can be cloned into retro- or lentiviral vectors that are used to transduce peripheral blood T cells from patients ex vivo, followed by expansion and infusion into patients (Fig. 1b). Typically, tumor antigen-specific T cells targeting selfantigens isolated from cancer patients are of low affinity, due to the impact of central tolerance on the T cell repertoire specific for these antigens. Attempts to overcome this issue have included the (i) engineering of high affinity TCRs by affinity maturation of the TCR [61], (ii) generation of murine TCRs by immunizing transgenic mice that express a HLA allele plus human tumor antigen [62], and (iii) isolation of TCRs in an allogeneic setting via in vitro induction of T cells specific for a foreign HLA-peptide complex [63], thereby bypassing the repertoire limitations imposed by thymic selection. In the first proof-of-principle study using genetically modified TCRs, T cells from metastatic melanoma patients were transduced with a TCR directed against the HLAA*0201/MART-1 peptide, which was cloned from a pure TIL culture isolated from a resected melanoma lesion of an HLA-*0201 patient that had responded to TIL treatment [31]. Sustained objective responses were observed in a minor proportion of the treated patients with no significant toxicity, and infused TCR-modified T cells were persistent for more than a year. Other trials have subsequently demonstrated significant and prolonged tumor regression in cancer patients using genetically modified TCRs against gp100 (melanoma) [32], NY-ESO-1 (melanoma, synovial sarcoma, multiple myeloma) [33], MAGE-A3 (myeloma, melanoma) [64], MAGE-A4 (esophageal cancer) [65], and CEA (colorectal carcinoma) [66] (Table 1). Fig. 2 Genetically modified T cells. (a) T cells recognize their target by the TCR complex, which is composed of the TCR α and β chain for recognition and the CD3 chains for signaling. T cells can be genetically engineered with defined specificity by expression of recombinant TCR αβ chains of known specificity. (b) CARs are composed of a single-chain fragment of variable region (scFv) derived from the antigen-binding domain of antibodies, fused to the CD3ζ transmembrane and intracellular signaling domains from the TCR complex. Additional intracellular signaling domains are added for costimulatory signals, such as the CD28 and 4-1BB signaling domains, to yield second- and third-generation CARs. Reprinted with permission from: Svane el al [19] Semin Immunopathol Although TILs have generally been safe, there are potential safety risks associated with the use of genetically modified T cell therapies, with the most critical being: (i) on-target offtumor toxicity, when infused T cells recognize normal tissue due to expression of the same antigen, such as gp100 and MART-1 which are expressed by both melanoma cells and normal melanocytes, (ii) off-target reactivity, when infused T cells cross-react against peptides other than those targeted, and (iii) cytokine-release syndrome (CRS), where infused T cells induce a sudden and dramatic increase of inflammatory cytokines [34, 67]. CAR-modified T cells The genetic modification of T cells with CARs combines the specificity of antibody-like recognition with the cytotoxic potency and activation potential of T cells (Fig. 2b). The construction of a CAR relies on the identification of a suitable antibody targeting a cell surface molecule of interest, and in contrast with the TCR modification approach, CAR recognition does not rely on peptide processing or presentation by MHC molecules. Thus, all surfaceexpressed target molecules represent a potential CARtriggering epitope. First-generation CARs are composed of an antigenbinding region (a single-chain antibody variable fragment (scFv)), based on the antibody of desired specificity, fused to the T cell signaling domains associated with native TCR signal transduction (Fig. 2b). These early CARs only provide activation signal 1 to T cells and have been shown to lead to CAR-T cell anergy upon repeated antigen stimulation [68]. Second generation CARs contain an additional co-stimulatory domain, such as CD28 or 4-1BB, which provides a second activation signal upon target antigen recognition (Fig. 2b). CAR-T cells carrying these CD28 or 4-1BB signaling moieties have demonstrated potent antitumor activity in clinical trials, resulting in meaningful clinical response rates that significantly outperform the previous generation (Table 1). Third generation CARs, which again incorporate another co-stimulatory domain (Fig. 2b), are now in development to further potentiate the persistence and activity of infused CAR-T cells. Multiple clinical trials have demonstrated the robust efficacy and frequent durable responses induced by CAR-T cells targeting CD19, a B cell-lineage antigen expressed on the surface of both normal and malignant B cells. CD19- specific CAR-T cells have been successfully used to treat patients with chemotherapy-refractory B cell malignancies, including marginal zone lymphoma, aggressive B cell lymphomas, chronic lymphocytic leukemia (CLL), and adult and pediatric acute lymphoblastic leukemia (ALL). In particular, the treatment of CLL and non-Hodgkin lymphoma resulted in tumor regressions for a majority of patients [38, 40, 41, 69, 70]; however, the most impressive results were observed in ALL, where complete response rates (CRR) of 70–90% in heavily pre-treated patients were regularly reported by several institutions testing CAR-T cell therapy [35–37, 71] (Table 1). Based on the collective experiences of these centers, which all utilized differing co-stimulatory domains and gene transfer methods, some key considerations can be identified: (i) patients should receive lymphodepleting chemotherapy, (ii) patients with acute lymphoid leukemia achieve very high response rates, (iii) off-tumor toxicity is primarily limited to B cell aplasia, a condition that can be clinically managed with prophylactic infusions of immunoglobulins, (iv) patients often develop severe CRS, and (v) there is no clear dose-response relationship between the number of CAR-T cells infused and the likelihood of response [35–39, 71–73]. Recent success among several groups exploiting CAR-T cell therapy targeting the B cell maturation antigen (BCMA) for the treatment of multiple myeloma, suggests that this modality may be extended to other hematological malignancies [42]. CAR-T cell therapy against solid tumors has yielded limited success thus far. Potential obstacles include (i) inefficient T cell localization to the tumor site, (ii) physical barriers preventing tumor infiltration by T cells, (iii) increased antigen selection difficulty due to the high antigen heterogeneity of solid tumors, (iv) high risk of ontarget, off-tumor toxicity due to the increased potential of target antigen expression in healthy essential organs, and (v) potent immunosuppressive factors that render T cells dysfunctional in the tumor microenvironment [74]. Ongoing preclinical research and clinical trials are attempting to overcome these obstacles by using modified gene transfer methods and treatment protocols, assessing novel CAR designs utilizing additional receptors and ligands to Barmor^ the CAR, and developing new targets such as CEA for colorectal cancers, disialoganglioside GD2 for neuroblastoma and sarcoma, PSMA for prostate cancer and melanoma, and EGFRvIII and IL13Rα2 for glioblastoma [74]. Summary The field of adoptive immunotherapy of cancer is a relatively new and rapidly expanding research area. Although immunotherapy techniques such as the adoptive transfer of tumorinfiltrating T cells and gene-modified T cells have been shown to mediate complete and durable responses in some patients with specific cancers, there are still many patients who derive no benefit from these therapies. However, there are many Semin Immunopathol promising ongoing research projects that may increase the number of patients that can benefit from this treatment modality and increase the feasibility of ACT as a standard of care treatment for all types of cancer. Compliance with ethical standards Conflict of interest The authors declare that they have no conflict of interest. Reference
Reference
-
1.Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736CrossRefPubMedGoogle Scholar
-
2.Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11:3887–3895PubMedPubMedCentralGoogle Scholar
-
3.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192:1027–1034CrossRefPubMedPubMedCentralGoogle Scholar
-
4.Hodi FS, O'Day SJ, McDermott DF et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723CrossRefPubMedPubMedCentralGoogle Scholar
-
5.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbé C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372:320–330CrossRefPubMedGoogle Scholar
-
6.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373:23–34CrossRefPubMedPubMedCentralGoogle Scholar
-
7.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L, KEYNOTE-001 Investigators (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372:2018–2028CrossRefPubMedGoogle Scholar
-
8.Sharma P, Callahan MK, Bono P, Kim J, Spiliopoulou P, Calvo E, Pillai RN, Ott PA, de Braud F, Morse M, le DT, Jaeger D, Chan E, Harbison C, Lin CS, Tschaika M, Azrilevich A, Rosenberg JE (2016) Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): a multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol 17:1590–1598CrossRefPubMedPubMedCentralGoogle Scholar
-
9.Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, Saba NF, Weiss J, Wirth L, Sukari A, Kang H, Gibson MK, Massarelli E, Powell S, Meister A, Shu X, Cheng JD, Haddad R (2017) Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-arm, phase II study. J Clin Oncol 35:1542–1549CrossRefPubMedPubMedCentralGoogle Scholar
-
10.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372:311–319CrossRefPubMedGoogle Scholar
-
11.Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348:56–61CrossRefPubMedGoogle Scholar
-
12.Rosenberg SA, Fox E, Churchill WH (1972) Spontaneous regression of hepatic metastases from gastric carcinoma. Cancer 29:472–474CrossRefPubMedGoogle Scholar
-
13.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 102:18538–18543CrossRefPubMedPubMedCentralGoogle Scholar
-
14.Loi S, Sirtaine N, Piette F, Salgado R, Viale G, van Eenoo F, Rouas G, Francis P, Crown JPA, Hitre E, de Azambuja E, Quinaux E, di Leo A, Michiels S, Piccart MJ, Sotiriou C (2013) Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol 31:860–867CrossRefPubMedGoogle Scholar
-
15.Clemente CG, Mihm MC Jr, Bufalino R et al (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310CrossRefPubMedGoogle Scholar
-
16.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pagès F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964CrossRefPubMedGoogle Scholar
-
17.Smith KA, Gilbride KJ, Favata MF (1980) Lymphocyte activating factor promotes T-cell growth factor production by cloned murine lymphoma cells. Nature 287:853–855CrossRefPubMedGoogle Scholar
-
18.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, Simpson C, Carter C, Bock S, Schwartzentruber D, Wei JP, White DE (1988) Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 319:1676–1680CrossRefPubMedGoogle Scholar
-
19.Svane IM, Donia M, Met Ö (2018) Adoptive T Cell Therapy. In: Haanen JBAG, Califano R, Lugowska I, Garassino MC (eds) Handbook of Immuno-Oncology, ESMO, Lugano, p 15–22Google Scholar
-
20.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA (2003) Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–342CrossRefPubMedPubMedCentralGoogle Scholar
-
21.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–2357CrossRefPubMedPubMedCentralGoogle Scholar
-
22.Andersen R, Borch TH, Draghi A, Gokuldass A, Rana MAH, Pedersen M, Nielsen M, Kongsted P, Kjeldsen JW, Westergaard MCW, Radic HD, Chamberlain CA, Hölmich LR, Hendel HW, Larsen MS, Met Ö, Svane IM, Donia M (2018) T cells isolated from patients with checkpoint inhibitor resistant-melanoma are functional and can mediate tumor regression. Ann Oncol 29(7):1575–1581Google Scholar
-
23.Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, Robbins PF, Rosenberg SA, Dudley ME (2008) Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother 31:742–751CrossRefPubMedPubMedCentralGoogle Scholar
-
24.Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM (2012) Characterization and comparison of ‘standard’ and ‘young’ tumour-infiltrating lymphocytes for adoptive cell therapy at a Danish translational research institution. Scand J Immunol 75:157–167CrossRefPubMedGoogle Scholar
-
25.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, Kubi A, Shalmon B, Hardan I, Catane R, Segal E, Markel G, Apter S, Nun AB, Kuchuk I, Shimoni A, Nagler A, Schachter J (2009) Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother 32:415–423CrossRefPubMedGoogle Scholar
-
26.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP (2005) Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 26:111–117CrossRefPubMedPubMedCentralGoogle Scholar
-
27.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854CrossRefPubMedPubMedCentralGoogle Scholar
-
28.Itzhaki O, Hovav E, Ziporen Y, Levy D, Kubi A, Zikich D, Hershkovitz L, Treves AJ, Shalmon B, Zippel D, Markel G, Shapira-Frommer R, Schachter J, Besser MJ (2011) Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J Immunother 34:212–220CrossRefPubMedGoogle Scholar
-
29.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, Glass M, Johnson VE, McMannis JD, Shpall E, Prieto V, Papadopoulos N, Kim K, Homsi J, Bedikian A, Hwu WJ, Patel S, Ross MI, Lee JE, Gershenwald JE, Lucci A, Royal R, Cormier JN, Davies MA, Mansaray R, Fulbright OJ, Toth C, Ramachandran R, Wardell S, Gonzalez A, Hwu P (2012) Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res 18:6758–6770CrossRefPubMedPubMedCentralGoogle Scholar
-
30.Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, Ho lmich LR, Hendel HW, Met O, Andersen MH, thor Straten P, Svane IM (2016) Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res 22:3734–3745CrossRefPubMedGoogle Scholar
-
31.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–129CrossRefPubMedPubMedCentralGoogle Scholar
-
32.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CCR, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–546CrossRefPubMedPubMedCentralGoogle Scholar
-
33.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee CCR, Levy CL, Li YF, el-Gamil M, Schwarz SL, Laurencot C, Rosenberg SA (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29:917–924CrossRefPubMedPubMedCentralGoogle Scholar
-
34.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, Kulikovskaya I, Sinha SK, Kronsberg S, Gupta M, Bond S, Melchiori L, Brewer JE, Bennett AD, Gerry AB, Pumphrey NJ, Williams D, Tayton- Martin HK, Ribeiro L, Holdich T, Yanovich S, Hardy N, Yared J, Kerr N, Philip S, Westphal S, Siegel DL, Levine BL, Jakobsen BK, Kalos M, June CH (2015) NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21:914–921CrossRefPubMedPubMedCentralGoogle Scholar
-
35.Davila ML, Riviere I, Wang X et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6:224ra25CrossRefPubMedPubMedCentralGoogle Scholar
-
36.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371:1507–1517CrossRefPubMedPubMedCentralGoogle Scholar
-
37.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, Qayed M, de Moerloose B, Hiramatsu H, Schlis K, Davis KL, Martin PL, Nemecek ER, Yanik GA, Peters C, Baruchel A, Boissel N, Mechinaud F, Balduzzi A, Krueger J, June CH, Levine BL, Wood P, Taran T, Leung M, Mueller KT, Zhang Y, Sen K, Lebwohl D, Pulsipher MA, Grupp SA (2018) Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 378:439–448CrossRefPubMedGoogle Scholar
-
38.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RPT, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, Raffeld M, Feldman S, Lu L, Li YF, Ngo LT, Goy A, Feldman T, Spaner DE, Wang ML, Chen CC, Kranick SM, Nath A, Nathan DAN, Morton KE, Toomey MA, Rosenberg SA (2015) Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33:540–549CrossRefPubMedGoogle Scholar
-
39.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, McSweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY (2017) Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med 377:2531–2544CrossRefPubMedPubMedCentralGoogle Scholar
-
40.Porter DL, Hwang WT, Frey NV et al (2015) Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7:303ra139CrossRefPubMedPubMedCentralGoogle Scholar
-
41.Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, Wood B, Lozanski A, Byrd JC, Heimfeld S, Riddell SR, Maloney DG (2017) Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol 35:3010–3020CrossRefPubMedGoogle Scholar
-
42.Brudno JN, Maric I, Hartman SD et al (2018) T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol 36(22):2267–2280Google Scholar
-
43.Garbe C (1993) Chemotherapy and chemoimmunotherapy in disseminated malignant melanoma. Melanoma Res 3:291–299PubMedGoogle Scholar
-
44.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y, Ohayon D, Shalmon B, Markel G, Yerushalmi R, Apter S, Ben-Nun A, Ben-Ami E, Shimoni A, Nagler A, Schachter J (2013) Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 19:4792–4800CrossRefPubMedGoogle Scholar
-
45.Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, Kudchadkar R, Zager J, Gibney G, Sondak VK, Weber J, Mulé JJ, Sarnaik AA (2012) Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother 35:615–620CrossRefPubMedPubMedCentralGoogle Scholar
-
46.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17:4550–4557CrossRefPubMedPubMedCentralGoogle Scholar
-
47.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, Zlott DA, Yang JC, Sherry RM, Kammula US, Klebanoff CA, Hughes MS, Restifo NP, Langhan MM, Shelton TE, Lu L, Kwong MLM, Ilyas S, Klemen ND, Payabyab EC, Morton KE, Toomey MA, Steinberg SM, White DE, Rosenberg SA (2016) Randomized, prospective evaluation comparing intensity of Lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol 34:2389–2397CrossRefPubMedPubMedCentralGoogle Scholar
-
48.Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, Salim M, Vallon-Christersson J, Törngren T, Kvist A, Ringnér M, Svane IM, Jönsson G (2017) Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat Commun 8:1738CrossRefPubMedPubMedCentralGoogle Scholar
-
49.Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH (2014) Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res 20:434–444CrossRefPubMedGoogle Scholar
-
50.Hilders CG, Ras L, van Eendenburg JD, Nooyen Y, Fleuren GJ (1994) Isolation and characterization of tumor-infiltrating lymphocytes from cervical carcinoma. Int J Cancer 57:805–813CrossRefPubMedGoogle Scholar
-
51.Yannelli JR, Hyatt C, McConnell S, Hines K, Jacknin L, Parker L, Sanders M, Rosenberg SA (1996) Growth of tumor-infiltrating lymphocytes from human solid cancers: summary of a 5-year experience. Int J Cancer 65:413–421CrossRefPubMedGoogle Scholar
-
52.Turcotte S, Gros A, Hogan K, Tran E, Hinrichs CS, Wunderlich JR, Dudley ME, Rosenberg SA (2013) Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: implications for adoptive cell transfer therapy. J Immunol 191:2217–2225CrossRefPubMedPubMedCentralGoogle Scholar
-
53.Andersen R, Westergaard MCW, Kjeldsen JW, Müller A, Pedersen NW, Hadrup SR, Met Ö, Seliger B, Kromann-Andersen B, Hasselager T, Donia M, Svane IM (2018) T-cell responses in the microenvironment of primary renal cell carcinoma-implications for adoptive cell therapy. Cancer Immunol Res 6:222–235CrossRefPubMedGoogle Scholar
-
54.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, Gartner J, Jia L, Trebska-McGowan K, Somerville RP, Robbins PF, Rosenberg SA, Goff SL, Feldman SA (2018) Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med 24:724–730CrossRefPubMedGoogle Scholar
-
55.Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, Gartner JJ, Zheng Z, Li YF, Ray S, Wunderlich JR, Somerville RP, Rosenberg SA (2015) Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350:1387–1390CrossRefPubMedGoogle Scholar
-
56.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA (2006) High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther 13:151–159CrossRefPubMedGoogle Scholar
-
57.Clay TM, Custer MC, Sachs J et al (1999) Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol 163:507–513PubMedGoogle Scholar
-
58.Tsuji T, Yasukawa M, Matsuzaki J, Ohkuri T, Chamoto K, Wakita D, Azuma T, Niiya H, Miyoshi H, Kuzushima K, Oka Y, Sugiyama H, Ikeda H, Nishimura T (2005) Generation of tumor-specific, HLA class I-restricted human Th1 and Tc1 cells by cell engineering with tumor peptide-specific T-cell receptor genes. Blood 106:470–476CrossRefPubMedGoogle Scholar
-
59.Peng PD, Cohen CJ, Yang S, Hsu C, Jones S, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA (2009) Efficient nonviral sleeping beauty transposon-based TCR gene transfer to peripheral blood lymphocytes confers antigen-specific antitumor reactivity. Gene Ther 16:1042–1049CrossRefPubMedPubMedCentralGoogle Scholar
-
60.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543:113–117CrossRefPubMedPubMedCentralGoogle Scholar
-
61.Robbins PF, Li YF, El-Gamil M et al (2008) Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 180:6116–6131CrossRefPubMedPubMedCentralGoogle Scholar
-
62.Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, Ruppert T, Bolhuis RLH, Melief CJ, Huber C, Stauss HJ, Theobald M (2001) Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol 2:962–970CrossRefPubMedGoogle Scholar
-
63.de Witte MA, Coccoris M, Wolkers MC, van den Boom M, Mesman EM, Song JY, van der Valk M, Haanen JB, Schumacher TN (2006) Targeting self-antigens through allogeneic TCR gene transfer. Blood 108:870–877CrossRefPubMedGoogle Scholar
-
64.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122:863–871CrossRefPubMedPubMedCentralGoogle Scholar
-
65.Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, Sugino S, Ueda S, Ishikawa T, Kokura S, Naota H, Ohishi K, Shiraishi T, Inoue N, Tanabe M, Kidokoro T, Yoshioka H, Tomura D, Nukaya I, Mineno J, Takesako K, Katayama N, Shiku H (2015) Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res 21:2268–2277CrossRefPubMedGoogle Scholar
-
66.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DAN, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA (2011) T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19:620–626CrossRefPubMedGoogle Scholar
-
67.Casucci M, Hawkins RE, Dotti G, Bondanza A (2015) Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 64:123–130CrossRefPubMedGoogle Scholar
-
68.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, Chen CC, Yang JC, Rosenberg SA, Hwu P (2006) A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12:6106–6115CrossRefPubMedPubMedCentralGoogle Scholar
-
69.Kalos M, Levine BL, Porter DL et al (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3:95ra73CrossRefPubMedPubMedCentralGoogle Scholar
-
70.Turtle CJ, Hanafi LA, Berger C et al (2016) Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8:355ra116CrossRefPubMedPubMedCentralGoogle Scholar
-
71.Park JH, Riviere I, Gonen M et al (2018) Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med 378:449–459CrossRefPubMedGoogle Scholar
-
72.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, Steinberg SM, Stroncek D, Tschernia N, Yuan C, Zhang H, Zhang L, Rosenberg SA, Wayne AS, Mackall CL (2015) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385:517–528CrossRefPubMedGoogle Scholar
-
73.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM, Robinson E, Steevens NN, Chaney C, Soma L, Chen X, Yeung C, Wood B, Li D, Cao J, Heimfeld S, Jensen MC, Riddell SR, Maloney DG (2016) CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126:2123–2138CrossRefPubMedPubMedCentralGoogle Scholar
-
74.Srivastava S, Riddell SR (2018) Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol 200:459–468CrossRefPubMedGoogle Scholar